Two-pass alignment of RNA-seq reads with STAR

Background
STAR 1-pass mapping mode aligns RNA-seq reads to the genome and generates alignment (SAM/BAM) and
splice junctions (SJ.out.tab) files. The 1-pass mapping mode generates all required data essential for many
downstream analyses such as differential gene expression analysis.
However, if your goal is to robustly and accurately identify novel splice junctions for differential splicing analysis and variant discovery, it is highly recommended to use the STAR in 2-pass mode.
In the two-pass mode, the genome indices are re-generated from splice junctions obtained from a 1-pass mode with the usual parameters and then run the mapping step (2-pass mapping). Optionally, you can also bypass genome indices re-generation and directly provide a 1-pass splice junction file during the 2-pass mapping step.
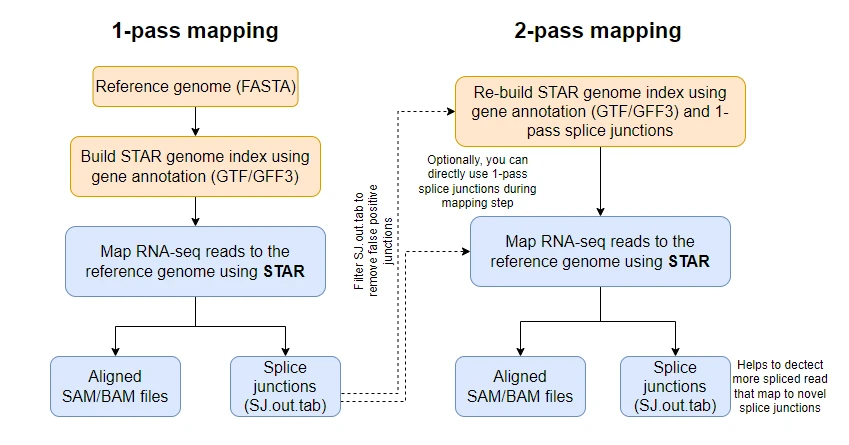
How to run STAR in 2-pass mode?
STAR can run in a 2-pass mode in two ways,
- By re-generating the genome indices from splice junctions obtained from 1-pass
- By using splice junctions directly during the mapping step
Re-generating the genome indices
Once you have successfully run the STAR in 1-pass mode and generated splice junctions (SJ.out.tab), you can use
these splice junctions for genome index re-generation.
Before using splice junctions, you should filter out the likely false positives splice junctions such as junctions supported
by very few reads (e.g. ≤ 2 reads), non-canonical junctions (The 0 value in column5 from SJ.out.tab denotes
non-canonical junctions), and annotated junctions (The 1 value in column6 from SJ.out.tab denotes
annotated junctions). The annotated junctions should be filtered out as they are already included in the gene annotation
(GTF/GFF3 file).
Below is the example splice junctions filtering command (you can modify it as per your requirement). The seed_sampleSJ.out.tab
is generated from running STAR in 1-pass mode.
cat seed_sampleSJ.out.tab | awk '($5 > 0 && $7 > 2 && $6==0)' | cut -f1-6 | sort | uniq > SJ_out_filtered.tab
# if you have multiple splice junctions files
cat *.tab | awk '($5 > 0 && $7 > 2 && $6==0)' | cut -f1-6 | sort | uniq > SJ_out_filtered.tab
Code source: https://groups.google.com/g/rna-star/c/Cpsf-_rLK9I
Non-canonical junctions may include a high number of false positive splice junctions and therefore it is recommended to filter them out. But if your study is interested in exploring the non-canonical junctions, you can keep them for 2-pass mapping.
Build genome index (with an additional parameter --sjdbFileChrStartEnd for filtered splice junctions),
# STAR version=2.7.10b
STAR --runThreadN 12 \
--runMode genomeGenerate \
--genomeDir ath_star_index_2pass \
--genomeFastaFiles Athaliana_TAIR10.fasta \
--sjdbGTFfile Athaliana_gene.gtf \
--sjdbFileChrStartEnd SJ_out_filtered.tab \
--sjdbOverhang 149
Once the genome indices are re-built, the single and paired-end RNA-seq reads can be mapped to the reference genome using STAR (2-pass mapping).
Genome mapping for single-end reads,
# STAR version=2.7.10b
# map single end reads to genome
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample.fastq \
--genomeDir ath_star_index_2pass \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix seed_sample \
--outSAMunmapped Within
Genome mapping for paired-end reads,
# map paired-end reads to genome
STAR --runThreadN 12 \
--readFilesIn ath_sample_read1.fastq ath_sample_read2.fastq \
--genomeDir ath_star_index_2pass \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix seed_sample \
--outSAMunmapped Within
If the read files are gzip compressed (*.fastq.gz), you can add an additional --readFilesCommand zcat or
--readFilesCommand gunzip -c parameter to the above mapping commands.
Direct 2-pass mapping
In this case, you can directly map the RNA-seq reads to the genome without re-building the genome indices (i.e. use genome indices created during 1-pass mode). You need to provide splice junctions obtained from 1-pass during the 2-pass mapping step. Please refer to my previous article on STAR 1-pass mapping for genome index building
Genome mapping for single-end reads,
# STAR version=2.7.10b
# map single end reads to genome
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample.fastq \
--genomeDir ath_star_index \
--sjdbFileChrStartEnd SJ_out_filtered.tab \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix seed_sample \
--outSAMunmapped Within
Genome mapping for paired-end reads,
# map paired-end reads to genome
STAR --runThreadN 12 \
--readFilesIn ath_sample_read1.fastq ath_sample_read2.fastq \
--genomeDir ath_star_index \
--sjdbFileChrStartEnd SJ_out_filtered.tab \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix seed_sample \
--outSAMunmapped Within
After successful 2-pass mapping, the STAR generates several output files (based on the input command) such as aligned BAM , mapping summary statistics, and log files for analyzing the mapping data.
You can refer to my previous article on STAR for detailed descriptions of output files
Enhance your skills with courses on genomics and bioinformatics
- Genomic Data Science Specialization
- Biology Meets Programming: Bioinformatics for Beginners
- Python for Genomic Data Science
- Bioinformatics Specialization
- Command Line Tools for Genomic Data Science
- Introduction to Genomic Technologies
References
- Schaarschmidt S, Fischer A, Zuther E, Hincha DK. Evaluation of seven different RNA-seq alignment tools based on experimental data from the model plant Arabidopsis thaliana. International journal of molecular sciences. 2020 Mar 3;21(5):1720.
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013 Jan 1;29(1):15-21.
- Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, Grant GR. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nature methods. 2017 Feb;14(2):135-9.
If you have any questions, comments or recommendations, please email me at reneshbe@gmail.com
This work is licensed under a Creative Commons Attribution 4.0 International License
Some of the links on this page may be affiliate links, which means we may get an affiliate commission on a valid purchase. The retailer will pay the commission at no additional cost to you.

