FASTQ Sequence Quality Format
What FASTQ Sequence Quality Format?
- FASTQ file (Sanger format) is a text file which represents a DNA/RNA sequence information in four lines, including sequence identifier (starts with @), DNA/RNA sequence (Nucleotide bases: A, T, G, C or Uncalled base: N), the sequence separator (usually + sign), and the sequence PHRED quality score encoded as ASCII characters (for space-efficient encoding)
- The quality scores are generated in binary base call (BCL) files from Illumina sequencing platforms, which are then
later converted to FASTQ files using
bcl2fastqtool
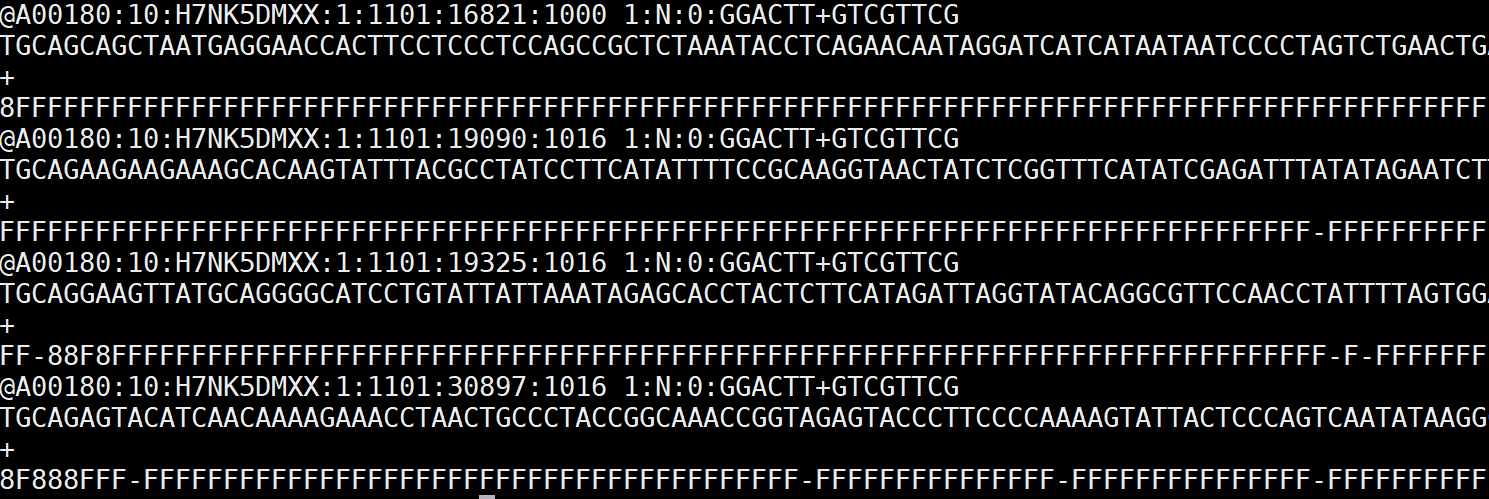
FASTQ sequence record for four sequences with quality encoding on fourth
line of each record (Awika et al., 2019)
-
The PHRED quality score \( Q_{PHRED} \) of an individual nucleotide base represents the error probability \( P_{e} \) of a given nucleotide base call is incorrect and it is given as,
\( Q_{PHRED} = -10 \log_{10}(P_{e}) \)
\( P_{e} = 10^{-Q_{PHRED}/10} \)
For example, if the \( Q_{PHRED} \) is 30, the \( P_{e} \) of a given base call will be 0.001. It means that there will be a 0.1 % chance that the given base call is wrong (99.9 % accuracy). The higher the quality score, the lesser the error in nucleotide base calling.
- FASTQ quality score encoding formats
| Format (type) | ASCII range | Q range |
|---|---|---|
| Sanger (PHRED) | 33 to 126 | 0 to 93 (typically 0 to 40) |
| Solexa/Illumina 1.2 or before (Solexa) | 59 to 126 | -5 to 62 (typically -5 to 40) |
| Illumina 1.3 and 1.4 (PHRED) | 64 to 126 | 0 to 62 (typically 0 to 40) |
| Illumina 1.5 to 1.7 (PHRED) | 64 to 126 | 0 to 62 (typically 3 to 40) |
| Illumina 1.8 and later (PHRED) | 33 to 126 | 0 to 93 (typically 0 to 41) |
Notes:
- Illumina 1.8 and later uses the same format as Sanger
- In Illumina 1.5 to 1.7, 0 and 1 quality scores are unused, while 2 (ASCII “B”) denotes Read Segment Quality Indicator and involved in read trimming
How to identify FASTQ Sequence Quality Format?
We will use bioinfokit v0.4 or later for checking the FASTQ Sequence Quality Format.
Check bioinfokit documentation for the latest version. After
installation, You can check the version using bioinfokit.__version__
After installing bioinfokit v0.4 or later, it can be used for detecting the FASTQ sequence quality format
I have used the subset of FASTQ dataset published in Bedre et al., 2019 for the analysis
# you can use interactive python interpreter, jupyter notebook, spyder or python code
# I am using interactive python interpreter (Python 3.7)
>>> from bioinfokit import analys, help
# Read documentation at https://github.com/reneshbedre/bioinfokit or
# get help message
>>> help.format.fq_qual_var()
# some examples
>>> analys.format.fq_qual_var(file="fq_18.fastq")
# output
# The fastq quality format is illumina 1.8+ (Offset +33)
# download dataset https://reneshbedre.github.io/assets/posts/fqformat/fq_sanger.fastq
>>> analys.format.fq_qual_var(file="fq_sanger.fastq")
# output
# The fastq quality format is Sanger (Offset +33)
References:
- Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic acids research. 2010 Apr 1;38(6):1767-71.
- Awika HO, Bedre R, Yeom J, Marconi TG, Enciso J, Mandadi KK, Jung J, Avila CA. Developing growth-associated molecular markers via high-throughput phenotyping in spinach. The Plant Genome. 2019 Nov 1;12(3).
- Bedre R, Irigoyen S, Schaker PD, Monteiro-Vitorello CB, Da Silva JA, Mandadi KK. Genome-wide alternative splicing landscapes modulated by biotrophic sugarcane smut pathogen. Scientific reports. 2019 Jun 20;9(1):1-2.
- https://en.wikipedia.org/wiki/FASTQ_format
If you have any questions, comments or recommendations, please email me at reneshbe@gmail.com
This work is licensed under a Creative Commons Attribution 4.0 International License

